How we built a graph neural network that finally sees the whole play — not just the audition
Every year, drugs that passed early safety tests go on to harm people in ways nobody predicted. Not because the chemistry was wrong. Not because the researchers were careless. But because we kept evaluating drugs the way a talent agent judges an actor from a solo audition tape.
Isolated. Out of context. No script. No co-stars. No stage.
In real theatre, a performance is never just about one actor. It depends on who they share the stage with, which scene they appear in, what the story demands at that moment. A brilliant performer in the wrong play, surrounded by the wrong cast, in the wrong context — can still wreck the whole production.
That is exactly how drug toxicity works. And that is exactly the problem we set out to fix with EIRION.
1. The First Attempt: Judging the Actor Alone
We started with a simple idea.
Take each compound, extract its chemical features, and train a model to predict toxicity. A classic approach — clean, efficient, proven.
So we built a Random Forest baseline.
In our analogy, this is like judging an actor purely based on their solo audition:
- No co-actors
- No storyline
- No context
Just the actor… alone.
Each compound got its own 1,024-bit Morgan fingerprint — a mathematical signature capturing the arrangement of atoms around every bond. Feed that into a Random Forest classifier, and it learns to associate certain chemical shapes with certain toxic outcomes.
And honestly? It worked. Our Random Forest hit a macro AUC of 0.7517 across 13 Tox21 bioassays. That is a solid number. Nothing to dismiss.
But something felt wrong.
Why the baseline falls short
The model made confident predictions — but not always the right ones.
We noticed it particularly around androgen receptor assays. The model was making decisions about drugs that act directly on hormone receptors, and it had no idea those receptors existed. The entire hormone system was invisible to it.
That made us stop and ask a harder question.
Why would a drug be toxic?
Not in the fingerprint sense. In the biological sense.
The real answer is: because it was blind to context. In reality, two very different compounds can cause the same toxicity if they affect the same gene. A compound’s behaviour can change entirely depending on which biological pathway it lands in. But our model could see none of that.
It was like saying: this actor is great, so the movie will be great — while ignoring who they are acting with, what story they are in, and how the scene unfolds.
Toxicity is not a property of a molecule in isolation. It is the result of a molecule stepping onto a very specific biological stage and disrupting the production that was already running.
And we were building models that had never even seen the stage.
2. The Insight: Biology Is a Story, Not a Snapshot
Here is what we knew about the biology our model was ignoring.
Drugs are not lone performers. They share the stage with genes — proteins that metabolise them, transport them, bind to them, activate or inhibit them.
Those genes themselves are part of pathways — larger storylines in the body’s ongoing biochemical production. The androgen receptor pathway. The cytochrome P450 metabolic pathway. The p53 DNA damage response.
When a drug enters the body, it immediately begins interacting with its supporting cast. CYP2D6 metabolises it. ABCB1 tries to pump it out. The androgen receptor either binds it or does not. These interactions — between compound and gene, between gene and pathway — determine whether the performance ends in applause or disaster.
The analogy maps cleanly:
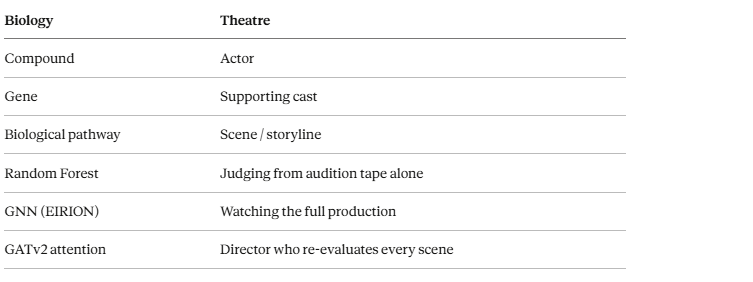
Biology Theatre Compound Actor Gene Supporting cast Biological pathway Scene / storyline Random Forest Judging from audition tape alone GNN (EIRION) Watching the full production GATv2 attention Director who re-evaluates every scene
The Random Forest had none of this. It was casting a play using only headshots.
We needed a model that could read the whole script.
3. Building the Stage: A Biological Knowledge Graph
That is when we shifted perspective entirely.
Instead of thinking about drugs as isolated data points, we built a graph — a network of relationships connecting every compound to the genes it interacts with, and every gene to the biological pathways it belongs to.
The graph has three node types and two edge types:
Nodes:
- Compounds (the actors, carrying their 1,024-bit Morgan fingerprint)
- Genes (the supporting cast)
- Pathways (the scenes and storylines)
Edges:
- Compound → Gene (this drug interacts with this cast member)
- Gene → Pathway (this cast member appears in this storyline)
With this structure, the question of toxicity stops being what does this molecule look like? and becomes how does this actor perform in this story, with this cast, in this scene?
That is a fundamentally richer question. And it demands a fundamentally richer model.
4. Why GNN: Learning from the Entire Play
To make use of this graph, we turned to Graph Neural Networks.
The key idea — called message passing — is worth understanding because it is genuinely elegant.
Imagine every actor in the theatre holding a notepad. At the start of rehearsal, they write down everything they know about themselves. Then, once per round, every actor passes their notepad to the people they share scenes with. They read their co-stars’ notes, incorporate that context, and update their own understanding.
After two rounds of this, each actor’s notepad contains not just their own identity — but everything they learned from their direct scene partners, and everything those partners learned from their co-stars.
That is message passing. After two GNN layers, a compound node in EIRION contains:
- Its own chemical fingerprint (what it looks like)
- Information from the genes it interacts with (who it shares the stage with)
- Information from the pathways those genes belong to (what storylines those cast members appear in)
Instead of asking “Is this actor good?” we now ask “How does this actor perform in this story, with this cast?”
That is a completely different question — and a far more powerful one.
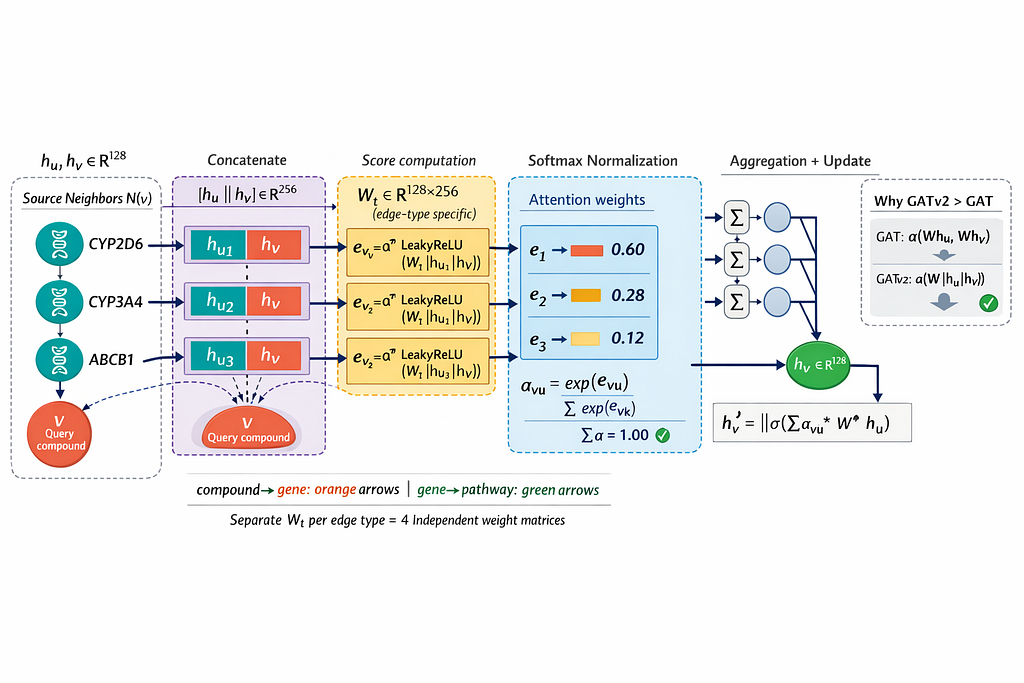
Why GATv2 specifically
Here is another reality about theatre: not every co-actor matters equally in every scene.
- In a romantic scene → the co-lead matters enormously
- In an action scene → the supporting characters might dominate
- In a solo monologue → others barely matter at all
The original GAT (version 1) behaved like a director who formed their opinion of each actor before rehearsals started — fixed, static, decided based on who the actors are rather than what they are actually doing together in the current scene.
GATv2 changes this. It computes attention based on the combined state of both nodes in each relationship simultaneously. The importance of a gene to a compound depends on the current state of both the compound and the gene at once — not a fixed score decided in advance.
For drug toxicity, this is crucial. CYP3A4 might be enormously important for metabolising one class of compounds and nearly irrelevant for another. GATv2 figures that out dynamically. GATv1 assigns it a fixed importance score and never revisits it.
The director who watches the actual performance always makes better casting decisions than the one who decided everything in advance.

5. Assembling the Production: Data & Architecture
Getting the idea right was one thing. Building the actual system meant assembling pieces from three major databases and confronting some genuinely messy data engineering problems.
The three data sources
Source What it provides Scale Role in EIRION Tox21 qHTS Compounds + assay labels 12,505 compounds · 13 assays Training data PharmGKB / CPIC Drug–gene relationships 25,041 genes · 21,298 edges Compound → gene edges Reactome Biological pathways 2,848 pathways · 48,593 edges Gene → pathway edges.
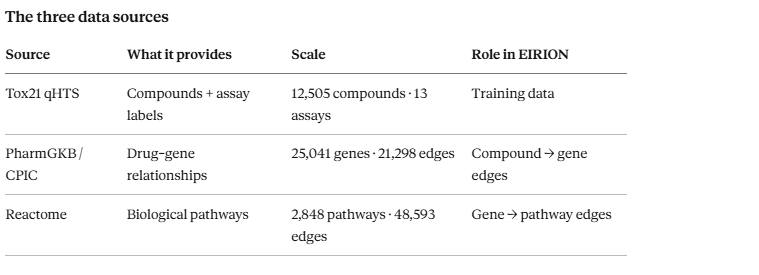
The dataset. We started with the Tox21 qHTS dataset — 12,505 compounds, each tested across 12 biological assays covering nuclear receptor signalling and stress response pathways. These are the assays that catch the kinds of toxicity that send drugs into withdrawal years after approval. Every compound came with a SMILES string (a text representation of its molecular structure) and binary labels indicating activity in each assay.
The cast. Gene data came from two sources: PharmGKB (the Pharmacogenomics Knowledgebase, maintained at Stanford) and CPIC (the Clinical Pharmacogenomics Implementation Consortium). Together these gave us 25,041 genes and 21,298 curated drug-gene relationships — direct evidence that specific drugs interact with specific genes, drawn from clinical studies and variant annotations.
The storylines. Pathway data came from Reactome, a curated biological pathway database. 2,848 human pathways with 48,593 edges connecting genes to the pathways they participate in.
The bridge problem
The datasets didn’t naturally align. PharmGKB identifies drugs using its own internal IDs. Our Tox21 compounds use NCGC registry IDs. Connecting them required computing InChIKeys — a standardised molecular hash — from InChI strings, then matching on the first 14 characters (the connectivity layer, which ignores stereochemistry).
This gave us 1,762 matches and 21,936 real compound-gene edges. For the remaining 11,568 compounds with no match, we connected them to 12 core pharmacogenomics toxicity genes — CYP2D6, CYP3A4, CYP2C19, and others — as a structural prior. Not ideal, but better than leaving most of the cast invisible. This is the biggest limitation of v1.0, and we address it directly in section 7.
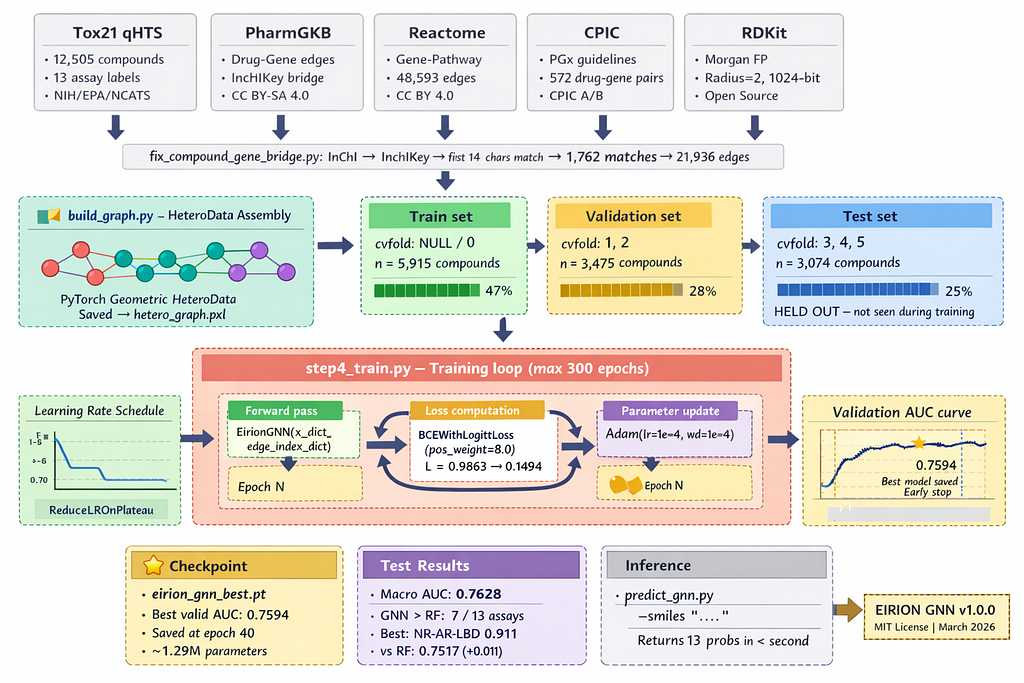
Model architecture
Two GATv2 layers with 4 attention heads each, hidden dimension of 128, dropout and layer normalisation after each layer, and residual connections to preserve the fingerprint signal throughout. A small MLP prediction head reads from compound embeddings and produces 13 binary toxicity scores — one per assay.
It is like letting the actors rehearse together, then evaluating their final performance as an ensemble — not as individuals.
We used ROC-AUC rather than accuracy as our primary metric, since toxic compounds represent only 2–10% of the data depending on the assay. A model predicting non-toxic for every compound would achieve over 90% accuracy on some assays — which is completely meaningless. We also weighted the loss function to penalise missing a true positive eight times more heavily than a false alarm. Missed toxicity has real consequences that false alarms do not.
6. Results: Did the Story Improve?
Yes — but in an interesting way.
The macro-average test AUC came in at 0.7628, compared to 0.7517 for the Random Forest baseline. An overall improvement of 1.5% across all 13 assays. But the macro average is not the whole story.
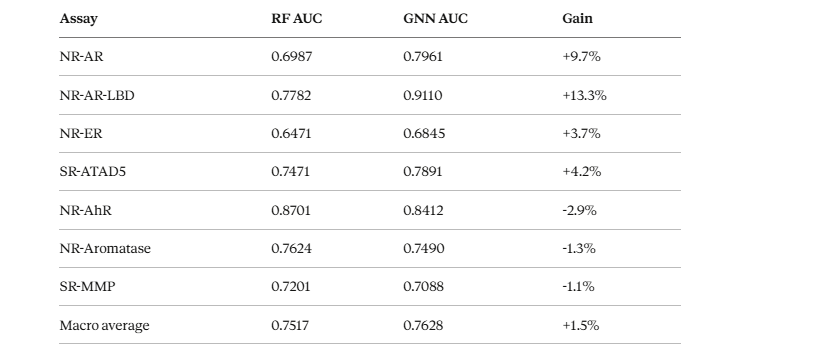
Where biological context wins
On the androgen receptor assays the improvements were dramatic — NR-AR-LBD improved by 13.3 percentage points. These assays measure whether a compound binds to the androgen receptor’s ligand binding domain. Of course a model that knows about the androgen receptor pathway and the genes involved performs better. The model that read the whole script understands this scene better than the model that only watched the audition tape.
Where Random Forest still holds its ground
Assays like NR-AhR, NR-Aromatase, and SR-MMP still favour the Random Forest. These are cases where the chemical fingerprint is a strong enough signal on its own, and where our compound-gene edge coverage happened to be thinner.
The pattern is clear and meaningful: graph context helps most when the assay is directly tied to a pathway that is well-represented in our biological graph. Context helps — but only when context exists.
7. The Honest Part: What EIRION Cannot Do Yet
We built something real, and it works. But part of building honestly is acknowledging what it cannot do yet.
Coverage is the biggest limitation. Only 937 of 12,505 compounds (but in EIRION v2 we are now covering over 55–72%) — about 7.5% — have genuine PharmGKB gene edges. The other 92.5% are connected to the 12 fallback PGx genes through a structural prior rather than actual biological evidence. For most of the graph, the model is still partially reading from an audition tape. The supporting cast is there, but most of the leads have not actually met them yet.
Gene and pathway features are sparse. Identity matrices rather than pre-trained biological embeddings mean each gene starts with almost no information about its actual biological role. The model has to learn gene representations from scratch through graph topology alone.
The model is transductive. It cannot score a brand new compound without rebuilding the entire graph. For a production system, that is not good enough.
These are the real constraints. We are not hiding them.
8. What Comes Next: Toward a Full Production
Version 2.0 — Better graph coverage
The plan is to expand compound-gene connections through three additional data sources: the Comparative Toxicogenomics Database, ChEMBL bioactivity data, and Tanimoto structural similarity inference for compounds that still lack real edges.
Projected coverage: from ~7.5% real compound-gene edges to 55–72%. That is not a marginal improvement. That is a completely different model.
Edge features will also carry metadata distinguishing pharmacokinetic relationships (the gene metabolises the drug) from pharmacodynamic ones (the gene is the drug’s direct target). Gene features will be replaced with Node2Vec embeddings trained on the gene-pathway co-membership graph, giving every gene a meaningful biological starting point.
Version 3.0 — Personalised medicine
This is the version that makes EIRION genuinely novel.
Every person carries variants in their CYP enzymes. Poor metabolisers process drugs more slowly. Ultrarapid metabolisers burn through them before they can work. If we encode a patient’s pharmacogenotype as features on the gene nodes, the model’s toxicity predictions stop being about the average person and start being about a specific individual.
Not: is this drug toxic? But: is this drug toxic for you, with your particular cast of metabolic enzymes?
That is personalised medicine. That is the version of this system with real clinical relevance.
Beyond individual drugs — multi-drug interactions
A patient on five medications is not running five independent plays. They are running one complex production where every actor affects every other actor. Extending EIRION with compound-compound edges from DrugBank’s drug-drug interaction dataset would let the model predict combinatorial toxicity — something no fingerprint-based classifier ever could do.
From “Is this drug toxic?” To “Is this drug toxic for this person, in this context, with these other drugs?”
That is the destination.
9. The Play Isn’t Over
We started with a simple idea: drug toxicity is not a solo performance.
We built a Random Forest that watches audition tapes, confirmed it works reasonably well, and then asked why it fails where it fails. The answer led us to build a knowledge graph spanning 12,505 compounds, 23,658 genes, 2,848 biological pathways, and nearly 50,000 curated biological edges.
We trained a Graph Attention Network that learns to watch the full performance — to weigh each cast member’s contribution based on the current context of the scene, not a fixed opinion formed before rehearsals began. We got a 13.3 percentage point improvement on the androgen receptor assay. We confirmed that biological context is not just philosophically important — it is mathematically measurable.
But what we built is still an early rehearsal. Most of our cast has not met their co-stars yet. The script has gaps. The director is still learning.
Somewhere right now, a compound that looks safe in isolation is stepping onto a biological stage it was never designed for. It will meet enzymes and receptors and pathways that change everything about its performance. And the model that only watched it audition alone will have no idea what is about to go wrong.
EIRION is our attempt to put a better director in the room. One that reads the whole script. One that watches the full production. One that understands — finally — that no actor, and no molecule, ever performs alone.
References
Datasets
- Tox21 Challenge. Tox21 quantitative high-throughput screening (qHTS) dataset. National Institutes of Health (NIH), U.S. Environmental Protection Agency (EPA), National Center for Advancing Translational Sciences (NCATS). Public domain. https://tox21.gov
- Whirl-Carrillo, M. et al. Pharmacogenomics knowledge for personalized medicine. Clinical Pharmacology & Therapeutics, 92(4), 414–417 (2012). PharmGKB: https://www.pharmgkb.org — Licensed CC BY-SA 4.0
- Caudle, K.E. et al. Standardizing terms for clinical pharmacogenetic test results. Clinical Pharmacology & Therapeutics, 100(4), 353–361 (2016). CPIC: https://cpicpgx.org — Licensed CC BY 4.0
- Jassal, B. et al. The Reactome pathway knowledgebase 2020. Nucleic Acids Research, 48(D1), D498–D503 (2020). Reactome: https://reactome.org — Licensed CC BY 4.0
Tools & Libraries
- Landrum, G. RDKit: Open-source cheminformatics software. (2006). https://www.rdkit.org
- Paszke, A. et al. PyTorch: An imperative style, high-performance deep learning library. Advances in Neural Information Processing Systems 32 (NeurIPS 2019). https://pytorch.org
- Fey, M. & Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. ICLR Workshop on Representation Learning on Graphs and Manifolds (2019). https://pyg.org
Key Papers
- Brody, S., Alon, U. & Yahav, E. How attentive are graph attention networks? International Conference on Learning Representations (ICLR 2022). https://arxiv.org/abs/2105.14491 (This is the GATv2 paper — the core architectural choice in EIRION)
- Veličković, P. et al. Graph attention networks. International Conference on Learning Representations (ICLR 2018). https://arxiv.org/abs/1710.10903 (Original GAT paper — what GATv2 improves upon)
- Mayr, A. et al. DeepTox: Toxicity prediction using deep learning. Frontiers in Environmental Science, 3, 80 (2016). (Foundational Tox21 deep learning benchmark)
- Rogers, D. & Hahn, M. Extended-connectivity fingerprints. Journal of Chemical Information and Modeling, 50(5), 742–754 (2010). (The Morgan fingerprint / ECFP paper — what the 1024-bit fingerprints are based on)
- Hamilton, W.L., Ying, R. & Leskovec, J. Inductive representation learning on large graphs. Advances in Neural Information Processing Systems 30 (NeurIPS 2017). https://arxiv.org/abs/1706.02216 (GraphSAGE — foundational message passing reference)
Want to Collaborate?
EIRION is an open project, and there is a lot of road still ahead.
If you are a researcher, bioinformatician, cheminformatician, or ML engineer who works in drug discovery, pharmacogenomics, or graph learning — I would genuinely love to hear from you. The problems that remain (coverage expansion, inductive inference, personalised PGx overlays, multi-drug interaction modelling) are hard and interesting, and they are better solved together than alone.
If any part of this project resonates with your work — whether that is the graph construction, the biological data engineering, the GATv2 architecture, or the clinical direction — reach out. I am open to collaboration, feedback, and expert input at any stage.
Author : Ajay Bharathi
Contact : ajayak0304@gmail.com
Tags : #GraphNeuralNetworks #GATv2 #MachineLearning #DeepLearning #DrugDiscovery #ToxicityPrediction #Bioinformatics #Eirion #PyTorch #DataScience #ExplainableAI #TechBlog
EIRION v1.0.0 is open source. The graph object, model checkpoint, and full training pipeline are available in the repository.
Published Mar 31, 2026 | 10 min read | Intermediate-Advanced
Why Drug Toxicity Can’t Be Predicted in Isolation — Building EIRION with Graph Neural Networks was originally published in Towards AI on Medium, where people are continuing the conversation by highlighting and responding to this story.